Questions:
38. What type of iris nodules are present in Neurofibromatosis Type 1, and Neurofibromatosis Type 2?
39. What are the ocular or CNS manifestations of Neurofibromatosis Type 1?
40. What are the ocular or CNS manifestations of Neurofibromatosis Type 2?
41. What is the classic findings triad in tuberous sclerosis?
42. What is the most prominent ocular manifestation of Tuberous sclerosis?
43. What autosomal dominant condition is associated with multiple, bilateral retinal angiomas and intracranial cerebellar hemangioblastomas?
44. What condition should be suspected in a patient with findings of telangiectasias of the conjunctival vessels and oculomotor apraxia?
45. A patient has an upper eyelid hemangioma, intraocular hypertension, and homonymous hemianopia, what condition is likely to be present?
46. What are the ocular manifestations of Sturge-Weber syndrome?
47. What are the findings of the Wyburn-Mason Syndrome?
48. What are the findings of the Klippel-Trenaunay-Weber Syndrome?
______________________________________________
Questions with answers:
38. What type of iris nodules are present in Neurofibromatosis Type 1, and Neurofibromatosis Type 2?
Neurofibromatosis Type 1 – Lisch nodules (iris pigment epithelium hamartomas)
Neurofibromatosis Type 2 – Lisch nodules and skin lesions are less common in NF2 than in NF1
39. What are the ocular or CNS manifestations of Neurofibromatosis Type 1?
1. Neurofibroma
2. Lisch nodules (iris pigment epithelium hamartomas)
3. Optic nerve glioma
4. Sphenoid dysplasia which may result in pulsatile exophthalmos or enophthalmos, orbital encephalocele, and orbital CSF.
40. What are the ocular or CNS manifestations of Neurofibromatosis Type 2?
1. Unilateral or Bilateral eighth cranial nerve masses
2. Neurofibroma
3. Meningioma
4. Glioma
5. Schwannoma
6. Juvenile posterior subcapsular cataract
7. Lisch nodules and skin lesions are less common in NF2 than in NF1
41. What is the classic findings triad in tuberous sclerosis (Bourneville disease)?
1. Adenoma sebaceum
2. Mental retardation
3. Epilepsy
Tuberous sclerosis is an autosomal dominant disorder, with high penetrance, and variable expressivity.
42. What is the most prominent ocular manifestation of tuberous sclerosis?
The most prominent ocular manifestation is a hamartoma of the retina and optic nerve. They are observed in up to 50% of patients but rarely cause visual loss. Seizures are the most common presenting symptom of tuberous sclerosis. Mental retardation occurs in more than 50% of these patients.
43. What autosomal dominant condition is associated with multiple, bilateral retinal angiomas and intracranial cerebellar hemangioblastomas?
Von-Hippel-Lindau disease is an autosomal dominant disorder with multiple, bilateral retinal angiomas and intracranial hemangioblastomas (most often in the cerebellum). Twenty-five percent of patients will have renal cell carcinoma. Five percent of patients will have a pheochromocytoma. The retinal angioma will typically have a feeder vessel, numerous exudates, and an exudative retinal detachment.
44. What condition should be suspected in a patient with findings of telangiectasias of the conjunctival vessels and oculomotor apraxia?
Ataxia telangiectasia is an autosomal recessive disorder with cerebellar ataxia, telangiectasias, immunodeficiency, and susceptibility to neoplasms. Classic ophthalmologic manifestations are telangiectasias of the conjunctival vessels and oculomotor apraxia.
45. A patient has an upper eyelid hemangioma, intraocular hypertension, and homonymous hemianopia, what condition is likely to be present?
Sturge-Weber Syndrome (encephalotrigeminal angiomatosis) which is characterized by cutaneous hemifacial hemangioma and hemangioma of the ipsilateral meninges and brain. It is a non-hereditary. In addition, there may be arteriovenous malformations, venous, and dural sinus abnormalities.
46. What are the ocular manifestations of Sturge-Weber syndrome?
a. Intraocular hypertension (increased episcleral venous pressure, immature angle, and neovascularization of the angle).
b. Glaucoma is particularly common when the hemangioma involves the upper eyelid.
c. Angiomas of the lid, conjunctiva, episclera, and uvea.
Associated findings include homonymous hemianopia contralateral to the meningeal hemangioma, seizures, headaches, and raised intracranial pressure.
47. What are the findings of the Wyburn-Mason Syndrome?
a. Retinal arteriovenous malformations with an intracranial arteriovenous malformation, typically in the ipsilateral brainstem.
B. It is nonhereditary.
48. What are the findings of the Klippel-Trenaunay-Weber Syndrome?
a. Large cutaneous hemangiomas with hypertrophy of the related bones and soft tissues
b. Retinal angiomas
______________________________________________
The information below is from: Neuro-ophthalmology Illustrated-2nd Edition. Biousse V and Newman NJ. 2012. Thieme
20.9 Phacomatoses
The phacomatoses are a group of disorders characterized by multiple hamartomas of the central and peripheral nervous systems, eye, skin, and viscera. The central nervous system lesions in phacomatoses have a very different natural history and prognosis than those found in other patients (e.g., the gliomas observed in the phacomatosis neurofibromatosis type 1 are often benign, whereas a glioma in a patient without neurofibromatosis may be a more aggressive tumor).
20.9.1 Neurofibromatosis Type 1
Neurofibromatosis type 1 (NF1; also known as von Recklinghausen disease) is the most common phacomatosis (1/5,000) and is autosomal dominant (NF1 gene localized on chromosome 17) with high penetrance and variable expressivity.
The diagnostic criteria for NF1 include two or more of the following:
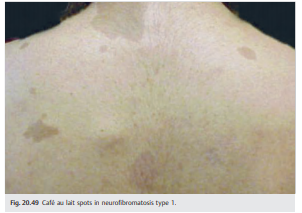
● Café au lait macules (≥ 6) (▶ Fig. 20.49)
● Neurofibromas (≥ 2) (▶ Fig. 20.50 and ▶ Fig. 20.51)
● Freckling (axillary, inguinal)
● Lisch nodules (iris pigment epithelium hamartomas) (▶ Fig. 20.52)
● Optic nerve glioma (▶ Fig. 20.53)
● Sphenoid dysplasia (▶ Fig. 20.54)
● First-degree relative with NF1
Café au lait macules (≥ 6) (▶Fig. 20.49)
Neurofibromas (≥2) (▶Fig. 20.50 and ▶Fig. 20.51)

Lisch nodules (iris pigment epithelium hamartomas) (▶Fig. 20.52)

Optic nerve glioma (▶Fig. 20.53)

Sphenoid dysplasia (▶Fig. 20.54)

The most prominent manifestation of NF1 is the involvement of cranial and peripheral nerves by two types of tumors:
● Schwannomas (neuromas, neurinomas, neurilemmomas)
○ Affect cranial nerves (fifth, third, fourth, and sixth nerves are most commonly affected)
● Neurofibromas
○ Plexiform neurofibromas
○ Localized neurofibromas
Central nervous system tumors (see ▶Fig. 20.53) are common in NF1.
● Optic nerve or chiasmal gliomas (in 15–20% of NF1 patients)
○ Often asymptomatic
○ May produce progressive loss of vision
○ May improve spontaneously
○ Treatment (chemotherapy, radiation) is performed only in cases with documented worsening of visual function.
● Low-grade astrocytic tumors
● Pulsatile exophthalmos or enophthalmos
● Herniation of dura, CSF, and brain into the orbit (encephalocele)
● May rarely result in compression of the extraocular muscles with diplopia and compression of the optic nerve with visual loss
Patients with NF1 may also have sphenoid dysplasia or absence of the sphenoid wing (see ▶Fig. 20.54).
20.9.2 Neurofibromatosis Type 2
Neurofibromatosis type 2 (NF2) is much less common than NF1 (1/50,000) and is an autosomal dominant disorder (NF2 gene localized on chromosome 22) with high penetrance.
The diagnostic criteria for NF2 include the following:
● Bilateral eighth cranial nerve mass identified on imaging or first degree relative with NF2 and
● Unilateral eighth nerve mass or two or more of the following:
○ Neurofibroma
○ Meningioma
○ Glioma
○ Schwannoma
○ Juvenile posterior subcapsular cataract
● Lisch nodules and skin lesions are less common in NF2 than in NF1.
● The most prominent manifestation of NF2 is bilateral acoustic neuromas (▶Fig. 20.55).

20.9.3 Tuberous Sclerosis (Bourneville disease)
Tuberous sclerosis is an autosomal dominant disorder, with high penetrance, and variable expressivity.
Patients present with a classic triad including the following:
● Adenoma sebaceum
● Mental retardation
● Epilepsy
Diagnosis
Definite diagnosis requires one primary feature and two secondary features or one secondary feature and two tertiary features.
● Primary features
○ Facial angiofibromas (▶Fig. 20.56)
○ Multiple ungual fibromas
○ Cerebral mass: cortical tubers, giant cell astrocytoma, calcified subependymal nodules protruding in the ventricle
○ Multiple retinal astrocytomas (▶Fig. 20.57)


● Secondary features
○ Affected first-degree relative
○ Cardiac rhabdomyosarcoma
○ Retinal hamartoma or retinal achromatic patch
○ Shagreen patch
○ Forehead plaque
○ Pulmonary lymphangiomyomatosis
○ Renal angiomyolipoma, renal cysts
● Tertiary features
○ Hypomelanotic macules
○ “Confetti” skin lesions
○ Renal, bone cysts
○ Nonrenal angiomyolipoma
○ Hamartomatous rectal polyps
○ Pulmonary lymphangiomyomatosis
○ Gingival lipomas
○ Infantile spasms
○ Cerebral white matter migration tracts or heterotopias
Seizures are the most common presenting symptom of tuberous sclerosis.
Mental retardation occurs in>50% of patients.
The most prominent ocular manifestation is hamartomas of the retina and optic nerve. They are observed in up to 50% of patients but rarely cause visual loss.
20.9.4 Von Hippel–Lindau Disease
Von Hippel–Lindau disease is an autosomal dominant disorder associating multiple, bilateral retinal angiomas and intracranial hemangioblastomas (most often in the cerebellum) (▶Fig. 20.58).

Twenty-five percent of patients will have a renal cell carcinoma. Five percent of patients will have a pheochromocytoma.
● Retinal angioma
○ Feeder vessel
○ Numerous exudates
○ Exudative retinal detachment
20.9.5 Ataxia Telangiectasia (Louis-Bar Syndrome)
Ataxia telangiectasia is an autosomal recessive disorder associating cerebellar ataxia, telangiectasias, immunodeficiency, and susceptibility to neoplasms.
Classic ophthalmologic manifestations include the following:
● Telangiectasias of the conjunctival vessels (▶Fig. 20.59)
● Oculomotor apraxia

20.9.6 Sturge-Weber Syndrome (Encephalotrigeminal Angiomatosis)
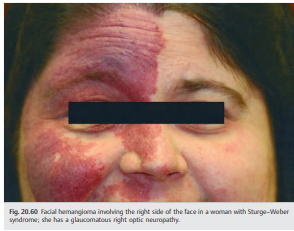
● Characterized by cutaneous hemifacial hemangioma associated with hemangioma of the ipsilateral meninges and brain (▶Fig. 20.60)
● Nonhereditary disorder
● Facial hemangioma present at birth (follows V1 and V2 innervation)
● Ocular manifestations ipsilateral to the hemangioma
○ Intraocular hypertension (increased episcleral venous pressure, immature angle, and neovascularization of the angle); glaucoma is particularly common when the hemangioma involves the upper eyelid.
○ Choroidal hemangioma
● Homonymous hemianopia contralateral to the meningeal hemangioma
● Seizures, headaches are common
● Raised intracranial pressure possible
20.9.7 Wyburn-Mason Syndrome
● Association of retinal arteriovenous malformations with an intracranial arteriovenous malformation, typically in the ipsilateral brainstem (see Fig. 20.17).
● Nonhereditary

20.9.8 Klippel-Trénaunay-Weber Syndrome
● Large cutaneous hemangiomas with hypertrophy of the related bones and soft tissues
● Retinal angiomas
Reference: 1. Neuro-ophthalmology Illustrated-2nd Edition. Biousse V and Newman NJ. 2012. Thieme
These questions are archived at https://neuro-ophthalmology.stanford.edu Follow https://twitter.com/NeuroOphthQandA to be notified of new neuro-ophthalmology questions of the week.
Please send feedback, questions, and corrections to tcooper@stanford.edu.