Questions:
27. Multiple sclerosis is more common in:
1. Men or Women?
2. African-Americans, Caucasians, or Hispanics?
28. What is Lhermitte sign and is it a classic finding in Multiple sclerosis?
29. What are the common eye symptoms of multiple sclerosis?
30. What is the 15-year risk of multiple sclerosis after an initial episode of optic neuritis:
1. overall
2. with no MRI lesions
3. with 1 MRI lesions
4. with 2 MRI lesions
5. with ≥ 3 MRI lesions
31. How common is mild-to-severe eye pain in optic neuritis?
32. What are the characteristics of Neuromyelitis Optica (Devic disease)?
33. What treatments are useful Neuromyelitis Optica?
________________________________________
Questions with answers?
27. Multiple sclerosis is more common in:
1. Men or Women?
2. African-Americans, Caucasians, or Hispanics?
Multiple sclerosis is more common in women and Caucasians.
28. What is Lhermitte sign and is it a classic finding in Multiple sclerosis?
Lhermitte sign is an electric shock-like sensation that runs down the back and into the limbs when the neck is flexed. The sign suggests a lesion of the dorsal columns of the cervical cord or of the caudal medulla. Although considered a classic finding in multiple sclerosis, it can be caused by a number of conditions.
29. What are the common eye symptoms of multiple sclerosis?
Visual symptoms and signs are very common and include the following:
● Optic neuritis is very common (often with Uhthoff Phenomenon)
○ Posterior (retrobulbar) in 65%
○ Anterior (with disc edema) in 35%
● Uveitis (most often pars planitis) is rare
● Retinal periphlebitis (asymptomatic)
● Visual field defects (optic tract lesion)
● Cranial nerve palsies (sixth, third, or fourth nerve palsy)
● Vertigo is very common
● Nystagmus is very common
● Internuclear ophthalmoplegia (bilateral>unilateral) is very common
● Skew deviation
30. What is the 15-year risk of multiple sclerosis after an initial episode of optic neuritis:
1. overall = 50%
2. with no MRI lesions = 25%
3. with 1 MRI lesions = 60%
4. with 2 lesions on MRI = 68%
5. with ≥ 3 lesions on MRI = 78%
Overall 50% developed MS
31. How common is mild-to-severe eye pain in optic neuritis?
In the ONTT, mild-to-severe eye pain present in 92% of patients.
32. What are the characteristics of Neuromyelitis Optica (Devic disease)?
The characteristics include:
1. Severe, immune-mediated demyelination and axonal damage, predominantly targeting optic nerves and the spinal cord
2. Associated with a serum NMO-IgG antibody that selectively binds aquaporin-4 (AQP4)
3. Presents as bilateral or rapidly sequential optic neuritis or transverse myelitis (often causing limb weakness and bladder dysfunction) or both with a typically relapsing course.
33. What treatment is useful Neuromyelitis Optica?
Acute attacks — All patients with suspected NMOSD should be treated for acute attacks. Suggested initial treatment is with high-dose intravenous methylprednisolone (1 gram daily for three to five consecutive days), in agreement with an expert panel, and based upon studies of multiple sclerosis and idiopathic optic neuritis.
For patients with severe symptoms, unresponsive to glucocorticoids, therapeutic plasma exchange is the suggested rescue treatment. Exchanges are carried out every other day up to a total of seven exchanges.
Limited retrospective and uncontrolled data suggest that patients with severe attacks of NMOSD do better if plasma exchange is started early as adjunctive therapy with glucocorticoids. One report of patients with severe attacks defined by an Expanded Disability Status Scale (table 4) score ≥4 and/or visual acuity ≤20/200) found that initial treatment with intravenous glucocorticoids plus early therapeutic plasma exchange was associated with improved outcomes compared with delayed initiation of plasma exchange following glucocorticoid treatment [122].
Attack prevention — Because the natural history of NMOSD is one of stepwise deterioration due recurrent attacks and accumulated disability, long-term immunotherapy is indicated for the prevention of attacks as soon as the diagnosis of NMOSD is made. For patients with NMOSD who are seropositive for aquaporin-4 (AQP4) IgG antibodies, we suggest treatment with eculizumab rather than another immunosuppressive agent. However, the optimal drug regimen and treatment duration are yet to be determined. Inebilizumab and satralizumab are not yet licensed or available outside of clinical trials.
Do Not use interferon beta drugs as this worsens the course of the disease
The information below is from: Neuro-ophthalmology Illustrated-2nd Edition. Biousse V and Newman NJ. 2012. Thieme
20.4 Multiple Sclerosis
Multiple sclerosis (MS) is a demyelinating disease affecting the white matter of the central nervous system (brain, optic nerves, and spinal cord). Young women are more commonly affected. The disease is more common in Caucasians than in other races.
20.4.1 Patient Evaluation
MS presents with relapsing-remitting neurological and visual symptoms and signs.
Common Symptoms and Signs of Multiple Sclerosis
The most common symptoms and signs of MS include the following:
● Optic neuritis (often with Uhthoff Phenomenon)
● Vertigo
● Trigeminal neuralgia
● Diplopia
● Sensory disturbances
● Cerebellar syndrome with ataxia
● Lhermitte sign (electric shock-like sensation down the back when flexing the neck)
● Spasticity and weakness
● Urinary incontinence
Symptoms are often multiple, and they come and go over days to weeks. There are no systemic manifestations of MS (no other organ than the central nervous system is affected). Headaches are uncommon as are seizures (only the white matter is typically affected). Altered mental status is uncommon.
Pearls
The association of neurologic signs with other organ failure, headaches, altered mental status, or seizures, should suggest a diagnosis other than MS, such as vasculitis.
Neuro-ophthalmic Manifestations of Multiple Sclerosis
Visual symptoms and signs are very common and include the following:
● Optic neuritis is very common
○ Posterior (retrobulbar) in 65%
○ Anterior (with disc edema) in 35%
● Uveitis (most often pars planitis) is rare
● Retinal periphlebitis (asymptomatic)
● Visual field defects (optic tract lesion)
● Cranial nerve palsies (sixth, third, or fourth nerve palsy)
● Vertigo is very common
● Nystagmus is very common
● Internuclear ophthalmoplegia (bilateral>unilateral) is very common
● Skew deviation
Pearls
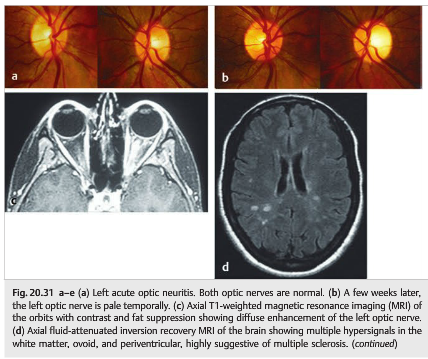
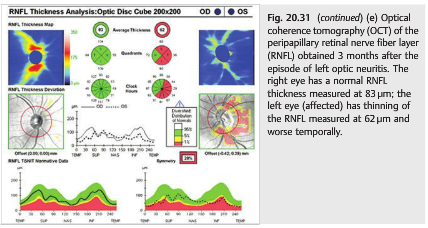
Optic neuritis is often the first sign of multiple sclerosis. Almost all patients with multiple sclerosis develop optic nerve involvement during the course of the disease (▶Fig. 20.31). Optical coherence tomography (OCT) is routinely used to follow patients with MS and is particularly useful in clinical trials (▶Fig. 20.31 e).
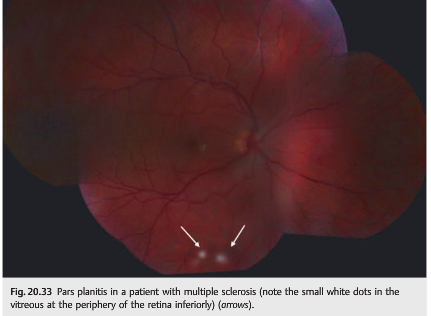
Peripheral venous sheathing and pars planitis are usually asymptomatic and found only when a careful dilated funduscopic examination is performed (▶Fig. 20.32 and ▶Fig. 20.33). They are not specific to MS, and they are often found in isolation or in patients with sarcoidosis.

20.4.2 Diagnosis
The diagnosis of MS is made when there is evidence of dissemination of the lesions in space (more than one type of deficit or more than one radiologic lesion) and time (relapse of events).
The diagnosis is suspected clinically and is confirmed by brain MRI. When patients present initially with a clinically isolated syndrome (such as optic neuritis), the clinical diagnosis of MS cannot be confirmed until a second clinical event occurs (clinically definite MS). ▶Table 20.3 presents the 2010 revised McDonald criteria for the diagnosis of MS.

20.4.3 Course of the Disease
A relapsing and remitting course is typical: the neurologic symptoms and signs of MS tend to improve spontaneously within a few weeks. They may resolve completely or improve only partially. Each relapse may be associated with a different type of deficit and may leave a residual deficit (neurologic deficit or visual loss) (relapsing-remitting form of MS).In the later course of the disease, the deficits (particularly weakness and spasticity)tend to progress slowly, without remission (secondary progressive form of MS). In a minority of cases, the deficit is slowly progressive from the beginning (primary progressive form of MS).
20.4.4 Treatment of Multiple Sclerosis-Related Neuro-ophthalmic Symptoms
Most MS patients develop visual symptoms at some point during the course of the disease. Monocular or binocular visual loss secondary to recurrent episodes of optic neuritis is common, and numerous patients experience binocular diplopia, often with oscillopsia related to nystagmus. The treatment of these neuro-ophthalmic manifestations of MS is similar to the one recommended for any neurologic flare of MS and is usually administered by the treating neurologist. Symptomatic treatment of persistent visual loss and diplopia is essential and requires a close collaboration with ophthalmologists (see Chapter 21). The treatment of pendular nystagmus is often challenging. It is important for MS patients to be followed by an ophthalmologist who will be able to confirm a diagnosis of optic neuritis when visual loss occurs (and will rule out other causes of visual loss).
The information below is from: Up-to-Date
Acute attacks — All patients with suspected NMOSD should be treated for acute attacks. We suggest initial treatment with high-dose intravenous methylprednisolone (1 gram daily for three to five consecutive days), in agreement with expert panel recommendations and based upon studies of multiple sclerosis and idiopathic optic neuritis [16,120,121]. (See “Treatment of acute exacerbations of multiple sclerosis in adults”, section on ‘Glucocorticoids’ and “Optic neuritis: Prognosis and treatment”, section on ‘Treatment’.).
For patients with severe symptoms, unresponsive to glucocorticoids, therapeutic plasma exchange is the suggested rescue treatment [16,120,121]. Exchanges are carried out every other day up to a total of seven exchanges.
Limited retrospective and uncontrolled data suggest that patients with severe attacks of NMOSD do better if plasma exchange is started early as adjunctive therapy with glucocorticoids. One report of patients with severe attacks (defined by an Expanded Disability Status Scale (table 4) score ≥4 and/or visual acuity ≤20/200) found that initial treatment with intravenous glucocorticoids plus early therapeutic plasma exchange was associated with improved outcomes compared with delayed initiation of plasma exchange following glucocorticoid treatment [122].
Seronegative NMOSD is managed in the same way as seropositive NMOSD.
Intravenous immune globulin has not been specifically evaluated for acute attacks of NMOSD and is rarely used in this setting.
Attack prevention — Because the natural history of NMOSD is one of stepwise deterioration due recurrent attacks and accumulated disability, long-term immunotherapy is indicated for the prevention of attacks as soon as the diagnosis of NMOSD is made [16,121,123]. For patients with NMOSD who are seropositive for aquaporin-4 (AQP4) IgG antibodies, we suggest treatment with eculizumab rather than another immunosuppressive agent. However, the optimal drug regimen and treatment duration are yet to be determined. Inebilizumab and satralizumab are not yet licensed or available outside of clinical trials.
● Eculizumab – Limited high-quality data support the effectiveness of eculizumab, a humanized antibody that binds to the complement component C5 and inhibits the formation of C5b-induced membrane attack complex. In a randomized controlled trial of 143 patients with NMOSD who were seropositive for aquaporin-4 (AQP4) IgG antibodies, treatment with eculizumab reduced the risk of relapse; the annualized relapse rates for the eculizumab and placebo groups were 0.02 and 0.35 (absolute risk reduction [ARR] 33 percent, rate ratio 0.04, 95% CI 0.01-0.15) [124]. Most patients in the trial were also taking concomitant immunotherapy (eg, azathioprine, glucocorticoids, mycophenolate mofetil) but not rituximab, which was excluded because its mechanism of action is incompatible with eculizumab.
Eculizumab is administered intravenously at 900 mg weekly for the first four doses, followed by maintenance dosing of 1200 mg every two weeks beginning at week five.
Note that eculizumab treatment is associated with an increased risk of infection with Neisseria meningitidis; patients should be immunized with meningococcal vaccines and receive daily antimicrobial prophylaxis, as described separately. In the United States, eculizumab is available only through a restricted program under a Risk Evaluation and Mitigation Strategy (REMS). (See “Treatment and prevention of meningococcal infection”, section on ‘Patients receiving C5 inhibitors’.)
Otherwise, eculizumab infusions are generally well tolerated. Commonly reported adverse events in clinical trials included headache, upper respiratory tract infections, back pain, and nausea.
● Inebilizumab – Inebilizumab is a humanized monoclonal antibody that binds to the CD19 surface antigen of B cells, thereby depleting a wide range of lymphocytes derived from B cell lineage. It was evaluated in the N-MOmentum trial of 230 patients with NMOSD (with 92 percent seropositive for anti-AQP4 antibodies) who were randomly assigned in a 3:1 ratio to intravenous treatment with inebilizumab 300 mg (n = 175) or to placebo (n = 56) administered on days 1 and 15 [125]. The trial was stopped early after an interim analysis at 6.5 months determined efficacy as defined by fewer attacks among patients assigned to inebilizumab compared with those assigned to placebo (12 versus 39 percent, ARR 27 percent, hazard ratio [HR] 0.27, 95% CI 0.15-0.50). The rate of all adverse events and serious adverse events was similar between the inebilizumab and placebo groups. Limitations of this trial include the short duration of clinical follow-up and early stopping, which can lead to an overestimate of the treatment effect. Further study is needed to establish the long-term efficacy and safety of inebilizumab relative to eculizumab, which has also shown efficacy in NMOSD.
● Satralizumab – Satralizumab is a humanized monoclonal antibody that binds interleukin-6 (IL-6) receptors, thereby suppressing inflammation mediated by IL-6 signaling pathways. Efficacy of satralizumab was demonstrated in a trial of 83 patients with NMOSD who were randomly assigned to subcutaneous injection with satralizumab 120 mg or placebo at weeks 0, 2, and 4, and every four weeks thereafter, in addition to their baseline immunosuppressant therapy [126]. At a median blinded treatment duration of 107 weeks, there were fewer relapses among patients assigned to satralizumab compared with those assigned to placebo (20 versus 43 percent, ARR 23 percent, HR 0.38, 95% CI 0.16-0.88). In the subgroup of 55 patients who were seropositive for anti-AQP4 antibodies, relapses were less frequent among patients assigned to satralizumab compared with placebo (11 versus 43 percent, ARR 32 percent, HR 0.21, 95% CI 0.06-0.75). In the subgroup of 28 patients who were seronegative for anti-AQP4 antibodies, the number of relapses was similar among patients assigned to satralizumab compared with placebo (36 versus 43 percent, ARR 7 percent, HR 0.66, 95% CI 0.2-2.24). For the entire cohort, the rate of all adverse events and serious adverse events was similar between the satralizumab and placebo groups.
● Other immunotherapies – The evidence of efficacy for systemic immunotherapy comes mainly from observational studies of agents including azathioprine [127,128], mycophenolate mofetil [129,130], rituximab [131], methotrexate [132,133], mitoxantrone [134], and oral glucocorticoids [135]. Among these, the agents most often considered as effective treatments for NMOSD are azathioprine, rituximab, and mycophenolate mofetil [120,136,137].
Comparative data for NMOSD treatments are scant.
• In an open-label randomized trial of 86 patients who had NMOSD with or without AQP4 antibodies, the reduction in the annualized relapse rate at 12 months was significantly greater for patients assigned to rituximab compared with those assigned to azathioprine [138].
• A retrospective, nonrandomized study from two tertiary centers in the United States analyzed relapses among patients with NMOSD who were treated with azathioprine and concomitant prednisone (n = 32) for at least six months, or with mycophenolate (n = 28) for at least six months, or with rituximab (n = 30) for at least one month, and followed up after treatment for at least six months [139]. Treatment with these agents was associated with significant reductions in annualized relapse rates in the range of 72 to 88 percent compared with baseline. As an example, the annualized relapse rate decreased from 2.26 before azathioprine treatment to 0.63 after treatment, a reduction of 72 percent. Treatment failure, defined as the development of any new central nervous system inflammatory event, varied from 33 to 53 percent.
Treatment with tocilizumab (an IL-6 receptor antagonist) has been associated with clinical stabilization or improvement in a small number of patients with refractory NMOSD who failed one or more of the “standard” treatments discussed above [140-144]. In an observational study of five Chinese patients with highly active NMOSD that was poorly responsive to “standard” immunosuppressive agents, treatment with bortezomib (a proteosome inhibitor that depletes plasma cells) was associated with relapse-free status in four and clinical stabilization in all five [145].
Limited observational evidence suggests that treatment of NMOSD with interferon beta, natalizumab, or fingolimod is not effective and may be harmful [146-151]. There is no published evidence regarding the treatment of NMOSD with ocrelizumab.
● Duration of therapy – Immunosuppression is usually continued for at least five years for patients who are seropositive for AQP4 antibodies, including those presenting with a single attack, because they are at high risk for relapse or conversion to NMOSD [152]. However, some experts suggest that life-long therapy is appropriate, given the often devastating nature of the disease. Others suggest that the length of immunotherapy should be tailored to the severity of attacks and disability.
References:
1. Neuro-ophthalmology Illustrated-2nd Edition. Biousse V and Newman NJ. 2012. Thieme
2. Multiple sclerosis risk after optic neuritis: final optic neuritis treatment trial follow-up. The Optic Neuritis Study Group. Arch Neurol. 2008 Jun; 65(6): 727–732.
Full Text https://www.ncbi.nlm.nih.gov/pmc/articles/PMC2440583/
3. Up-to-Date Neuromyelitis optica spectrum disorders
Authors: Christopher C Glisson, DO, MS, FAAN
Section Editor: Francisco González-Scarano, MD
Deputy Editor: John F Dashe, MD, PhD
Literature review current through: June 2020. | This topic last updated: Dec. 11, 2019.
These questions are archived at https://neuro-ophthalmology.stanford.edu
Follow https://twitter.com/NeuroOphthQandA to be notified of new neuro-ophthalmology questions of the week.
Please send feedback, questions, and corrections to tcooper@stanford.edu.