Questions:
8. What tests should be done in all patients over age 50 with transient or permanent diplopia?
9. What is the Silent Sinus Syndrome?
10. What is the Kearns-Sayre syndrome?
11. What are 5 mitochondrial diseases that can have ophthalmoplegia?
12. What are the features of Myotonic Dystrophy?
13. What are the features of Oculopharyngeal Dystrophy?
14. What 3 conditions other than paresis or restriction should be considered in any adult with new-onset transient or permanent ocular misalignment?
15. What may induce the myasthenia gravis or make it worse?
16. What is the mechanism of action of edrophonium?
17. What are the side effects of the Tensilon test?
18. What does the development of lid retraction in a patient with Myasthenia suggest?
19. What percent of patients presenting with purely ocular myasthenia will progress to develop generalized disease?
20. Does myasthenia gravis ever affect the pupils?
21. Does myasthenia gravis ever cause pain?
Questions with answers:
8. What tests should be done in all patients over age 50 with transient or permanent diplopia?
ESR, CRP, CBC and platelets. At least 10% of patients with permanent visual loss from giant cell arteritis report having episodes of transient diplopia within the weeks preceding loss of vision.
9. What is the Silent Sinus Syndrome?
Silent sinus syndrome is a condition characterized by enophthalmos and/or hypoglobus secondary to the collapse of the orbital floor in the presence of asymptomatic chronic maxillary sinusitis.
10. What is the Kearns-Sayre syndrome?
The Kearns-Sayre syndrome is a subset of chronic progressive external ophthalmoplegia (CPEO) with additional neurologic and systemic abnormalities. Clinical diagnostic criteria are:
1. Onset prior to age 20,
2. CPEO,
3. Retinal pigmentary degeneration, and at least one of the following:
a. Cardiac conduction abnormalities,
b. Elevated cerebrospinal fluid protein (greater than 100 mg/dL), or
c. Cerebellar dysfunction. Muscle biopsy shows ragged red fibers. It has a single, large deletion of mitochondrial DNA.
11. What are 5 mitochondrial diseases that can have ophthalmoplegia?
1. Kearns-Sayre syndrome
2. MELAS syndrome (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes)
3. MNGIE syndrome (mitochondrial neurogastrointestinal encephalomyopathy)
4. SANDO syndrome (sensory ataxic neuropathy, dysarthria, and ophthalmoplegia)
5. Leigh syndrome (subacute necrotizing encephalomyelopathy).
12. What are the features of Myotonic Dystrophy?
1. Bilateral ptosis with progressive ophthalmoplegia
2. Typical “Christmas tree” cataract
3. Progressive weakness of distal muscles with myotonia (involuntary delayed relaxation following contraction, such as after sustained grip-cannot let your hand go)
4. Typical facies with frontal balding, facial weakness, long face, temporal wasting
5. Cardiac conduction defects
6. Most common adult-onset muscular dystrophy
7. Autosomal dominant (trinucleotide repeat on chromosome 19)
8. Age of onset is younger in successive generations is common
13. What are the features of Oculopharyngeal Dystrophy?
1. Bilateral ptosis with progressive ophthalmoplegia
2. Dysphagia
3. Presents late in life
4. More common in French Canadians.
14. What 3 conditions other than paresis or restriction should be considered in any adult with new onset transient or permanent ocular misalignment?
1. Myasthenia (ocular or generalized)
2. Wernicke encephalopathy both of which can mimic any combination of extraocular movement dysfunction with normal pupils,
3. If over age 50, cranial arteritis.
15. What may induce the myasthenia gravis or make it worse?
This autoimmune condition may be induced by medications affecting neuromuscular transmission (curare, penicillamine). It is often worsened by medications (curare, aminoglycosides, penicillamine, beta-blockers, benzodiazepines). Rarely it is a paraneoplastic disorder.
16. What is the mechanism of action of edrophonium?
Edrophonium (Tensilon) is a short-acting acetylcholinesterase inhibitor that increases the concentration of acetylcholine at the synapse and, therefore, temporarily reverses signs of myasthenia gravis.
17. What are the side effects of the Tensilon test?
Tensilon (Edrophonium) is given intravenously. Its effects last 2 to 3 minutes. Side effects include sweating, fasciculations, diarrhea, abdominal cramps, palpitations, or bradycardia. Although it is generally well tolerated, hypotension, vagal reactions, and even respiratory or cardiac arrest are possible. It should be avoided in patients with known respiratory or cardiac diseases and in the elderly.
18. What does the development of lid retraction in a patient with Myasthenia suggest?
Thyroid eye disease. Thyroid eye disease is the autoimmune disorder most commonly associated with ocular myasthenia. Each disease occurs more frequently in patients with the other disease than in the general population.
19. What percent of patients presenting with purely ocular myasthenia will progress to develop generalized disease?
Between 50 and 60% of patients presenting with purely ocular myasthenia will progress to develop generalized disease, and the vast majority of those who do will do so within1 to 2 years.
20. Does myasthenia gravis ever affect the pupils?
In myasthenia gravis, the pupils are always normal.
21. Does myasthenia gravis ever cause pain?
In myasthenia gravis, the pupils are always normal and there is no pain.
________________________________________________
The information below is from: Neuro-ophthalmology Illustrated-2nd Edition. Biousse V and Newman NJ. 2012. Theme
Trauma
Orbital trauma can restrict free movements of the globe in the orbit and often results in diplopia. Sudden orbital compression (e.g., tennis ball hitting the eye) can produce a blowout fracture of the orbital floor, entrapping the inferior rectus and other orbital tissue in the fracture. Elevation of the eye is restricted by the entrapped muscle, and depression of the eye is also generally poor because the muscle cannot constrict normally (▶Fig. 13.43).
Orbital fracture can also result in contusion of the extraocular muscle; hemorrhage in the extraocular muscle, with subsequent fibrosis; or orbital hemorrhage. Acute (and also sometimes delayed) enophthalmos and inferior orbital nerve damage (responsible for hypoesthesia and numbness of the inferior orbital rim and of the ipsilateral cheek) commonly accompany orbital floor fractures. A medial blowout fracture is also possible and results in entrapment of the medial rectus with an abduction deficit that may mimic a sixth nerve palsy (▶Fig. 13.52).
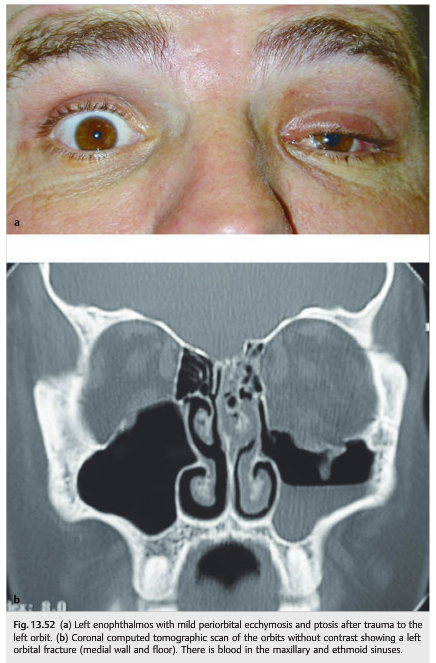
Other mechanisms of diplopia after head trauma include trauma to a cranial nerve (fourth nerve palsy most common, followed by sixth and third nerve palsies), carotid-cavernous fistula, brainstem lesion, subarachnoid hemorrhage with cranial nerve palsies, and raised intracranial pressure with sixth nerve palsy.
Giant Cell Arteritis and Orbital Ischemia
Chronic orbital ischemia (e.g., patient with common or internal carotid occlusion with poor collateral circulation, or patient with systemic vasculitis such as giant cell arteritis) can result in ischemia of an extraocular muscle, thereby producing diplopia, usually with pain. The diplopia is often transient and can be horizontal or vertical. At least 10%of patients with permanent visual loss from giant cell arteritis report having episodes of transient diplopia within the weeks preceding loss of vision (see Chapter 20).
Pearls
Giant cell arteritis needs to be ruled out in all elderly patients with transient or permanent diplopia.
Bony Deformation of the Orbit
Bony deformations change the shape of the orbit and also change the orientation of the orbital walls. In addition to compressing intraorbital structures, these changes modify the way the extraocular muscles move in the orbit and often produce diplopia.
Fibrous Dysplasia
Fibrous dysplasia is a benign bone condition in which normal bone is replaced by immature bone and osteoid in a cellular fibrous matrix. This results in expansion of the bone, further resulting in pain; deformation of the face, orbit, and cranium; and compression of adjacent structures (▶Fig. 13.53).
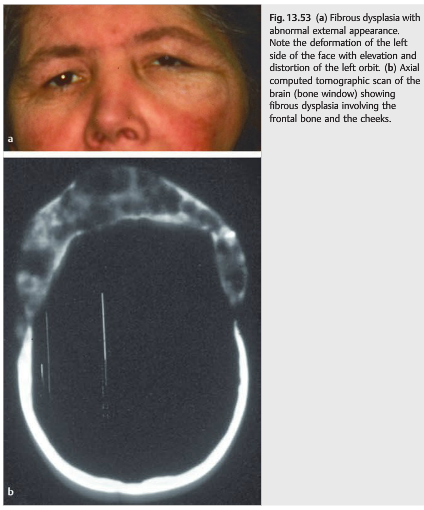
Diplopia is common secondary to displacement of the globe and of the ocular muscles, as well as cranial nerve palsies. Associated visual loss happens when there is compression of the optic nerve (usually at the level of the optic canal).Surgical debulking is often performed to improve the craniofacial deformity and to relieve cranial nerve compression. Surgical decompression of the orbital apex and optic canal is performed when there is a progressive optic neuropathy.
Agenesis of the Sphenoid Wing
Agenesis of the sphenoid wing occurs classically in the setting of neurofibromatosis type 1. It results in herniation of the intracranial contents into the orbit (▶Fig. 13.54).
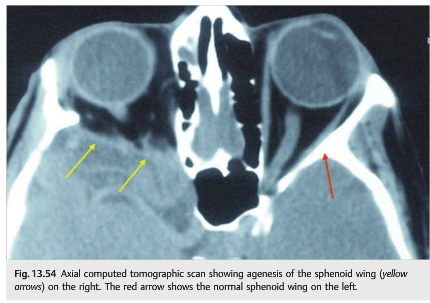
Clinically, patients present with the following:
● Pulsatile proptosis (pulsations of the cerebrospinal fluid push directly on the eye)
● Variable diplopia
● Various degrees of visual loss when the optic nerve is compressed
If the visual function is normal, there is usually no treatment necessary.
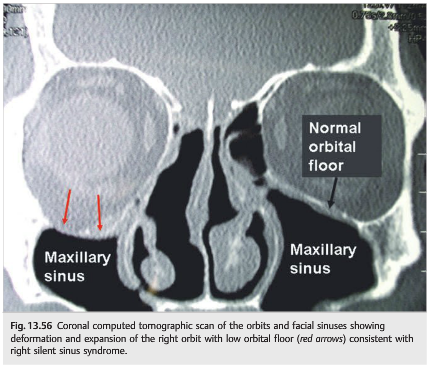
Silent Sinus Syndrome
Patients who have chronic maxillary sinusitis develop atrophy of the maxillary sinus with resultant lowering of the ipsilateral orbital floor (which constitutes the superior wall of the maxillary sinus). This may result in displacement of the orbital contents and ipsilateral hypoglobus, often with an elevation deficit of the eye. These patients may have binocular vertical diplopia, worse in up gaze (▶Fig. 13.55 and ▶Fig. 13.56).
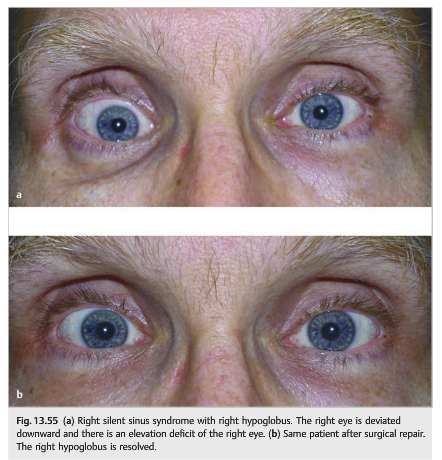
Progressive Myopathies Commonly Affecting the Extraocular Muscles
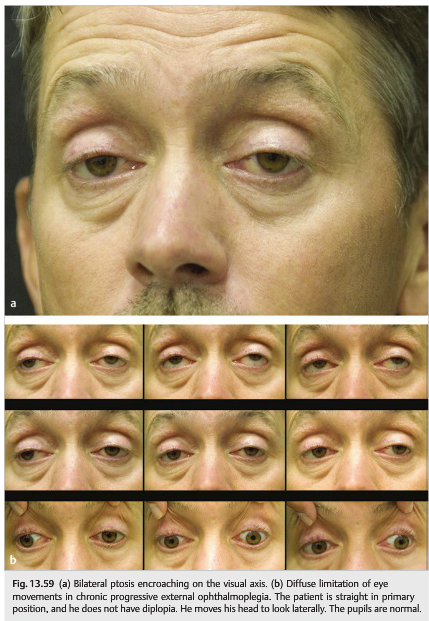
The syndrome of chronic progressive external ophthalmoplegia (CPEO) is characterized by progressive limitation of eye movements and ptosis (often over many years). The pupils are always normal. Commonly, patients do not complain of diplopia because their eyes remain relatively straight in primary position. The most common first complaint of diplopia occurs with reading because of convergence insufficiency.
Several disorders can present with CPEO, and often there are other neurologic signs such as diffuse facial and limb myopathy. In general, imaging is normal, and the extraocular muscles are not enlarged. Myopathies affecting the extraocular muscles are often associated with systemic myopathy and cardiomyopathy. Therefore, all patients with ocular myopathy need to have an electrocardiogram. Finding a cardiac conduction block may be life-saving. Myasthenia gravis needs to be ruled out
Mitochondrial Myopathies
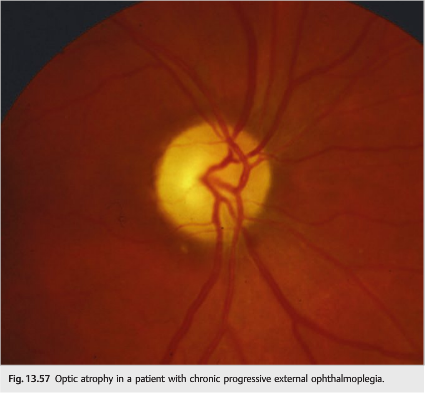
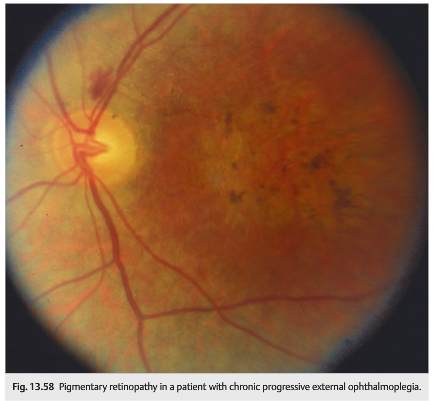
In mitochondrial disease, CPEO can occur any time from infancy to old age, and the onset is typically insidious. Ophthalmoplegia and ptosis may be isolated or associated with other neurologic or systemic abnormalities (▶Fig. 13.57, ▶Fig. 13.58, and ▶Fig. 13.59)
● Common neurologic findings: facial, bulbar, and limb myopathies, deafness, ataxia, spasticity, peripheral neuropathy, gastrointestinal myopathy and neuropathy, vestibular dysfunction, dementia, episodic encephalopathy or coma, and calcification of the basal ganglia
● Associated ocular features: optic atrophy, pigmentary retinopathy, corneal opacities, corneal edema, and cataracts
● Systemic manifestations: involve the cardiac, endocrine, skin, or skeletal systems and include cardiac conduction abnormalities, short stature, diabetes mellitus, delayed sexual maturation, hypogonadism, hypomagnesemia, hypoparathyroidism, hypothyroidism, and respiratory insufficiency.
The Kearns–Sayre syndrome is a subset of CPEO in which neurologic and systemic abnormalities figure most prominently.
Clinical diagnostic criteria include the following:
● Onset prior to age 20
● CPEO
● Retinal pigmentary degeneration
● At least one of the following:
○ Cardiac conduction abnormalities
○ Elevated cerebrospinal fluid protein (> 100 mg/dL)
○ Cerebellar dysfunction
The other neurologic and systemic abnormalities associated with mitochondrial myopathies (as already described) occur commonly in patients with Kearns–Sayre syndrome.
Muscle biopsy shows ragged red fibers (demonstrated on modified trichrome stain); they can be seen in limb and extraocular muscles in nearly all cases of Kearns–Sayre syndrome and in some patients with isolated CPEO.
Mitochondrial DNA analysis of skeletal muscle tissue of some CPEO patients reveals rearrangement of segments of mitochondrial DNA in the form of deletions and duplications.
Other Mitochondrial Diseases That Can Have Ophthalmoplegia
Myopathy involving the extraocular muscles is common in most mitochondrial diseases.
● MELAS syndrome (mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes)
● MNGIE syndrome (mitochondrial neurogastrointestinal encephalomyopathy)
● SANDO syndrome (sensory ataxic neuropathy, dysarthria, and ophthalmoplegia)
● Leigh syndrome (subacute necrotizing encephalomyelopathy)
Myotonic Dystrophy (▶Fig. 13.60)
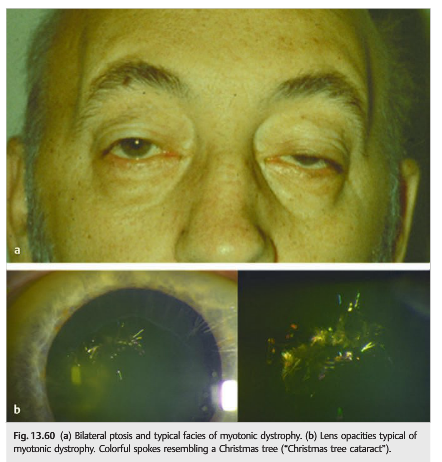
● Most common adult-onset muscular dystrophy
● Autosomal dominant (trinucleotide repeat on chromosome 19)
● Anticipation common (age of onset is younger in successive generations)
● Bilateral ptosis with progressive ophthalmoplegia
● Characteristic cataract (“Christmas tree”)
● Progressive weakness of distal muscles with myotonia (involuntary delayed relaxation following contraction, such as the inability to let go after a sustained grip)
● Typical facies with frontal balding, facial weakness, long face, temporal wasting
● Cardiac conduction defects
Oculopharyngeal Dystrophy
● Autosomal dominant (trinucleotide repeat on chromosome 14)
● More common in French Canadians
● Presents late in life
● Bilateral ptosis with progressive ophthalmoplegia
● Dysphagia
Congenital and Developmental Disorders Affecting the Extraocular Muscles
Numerous congenital abnormalities affecting the extraocular muscles may result in eye movement abnormalities in young children.
Agenesis of the Extraocular Muscles
Congenital Agenesis of the extraocular muscles is rare; most cases involve only one muscle.
Anomalies of Extraocular Muscle Origin and Insertion
The following disorders are responsible for various abnormalities of eye movement in young children.
Brown Syndrome
Patients with Brown syndrome have an elevation deficit of the affected eye secondary to restriction of the superior oblique tendon. The eye does not elevate when the eye is in adduction (there is a downshoot of the eye in adduction). Brown syndrome may be congenital or acquired.
Congenital Brown syndrome (▶Fig. 13.61)
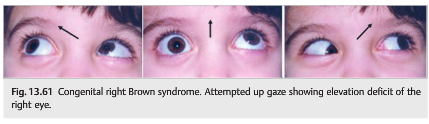
● Arises from congenitally short or inelastic tendon of the superior oblique muscle resulting in mechanical restriction of elevation of the eye when adducted
● Is commonly associated with normal binocular vision without amblyopia
Acquired Brown syndrome
● Can occur as a result of orbital trauma, an orbital inflammatory or infectious process, or after orbital injections
● Is often associated with pain at the level of the trochlea (superior medial corner of the orbit)
● Can result from rheumatoid arthritis
Congenital Adherence of Extraocular Muscles
● Adhesion between the sheaths of the lateral rectus and inferior oblique muscles, causing deficient abduction of the eye (usually bilateral)
● Adhesion between the sheaths of the superior rectus and superior oblique muscles, causing deficient elevation of the eye
Congenital Myopathies
Congenital myopathies make up a group of congenital systemic myopathies also affecting the extraocular muscles and causing ptosis and bilateral ophthalmoplegia.
● Myotubular myopathy (centronuclear myopathy)
● Nemaline myopathy
● Central core myopathy
● Multicore myopathy
Congenital Fibrosis of the Extraocular Muscles (CFEOM)
● Rare cause of ocular motility restriction, usually familial
● Presents in childhood
● Any individual extraocular muscle, or combination of muscles, may be replaced by fibrotic tissue, resulting in variable degrees of ophthalmoplegia and ptosis. The Syndrome of CFEOM is associated with anomalies of the cranial nerves and their nuclei and is part of the congenital cranial dysinnervation disorders, similar to Duane Syndrome, Möbius syndrome, and congenital facial palsy.
● Amblyopia is common.
13.5.2 The Lesion Is at the Neuromuscular Junction
Ocular Myasthenia and Myasthenia Gravis
Myasthenia gravis is essentially the only disease to clinically affect extraocular neuromuscular junction transmission. It presents with isolated ptosis or diplopia in 50% of cases.
● Autoimmune disorder
● Autoantibodies directed against the postsynaptic acetylcholine receptors blocked destroy the receptors. This decreased number of functional acetylcholine receptors results in deficient synaptic transmission at the level of the junction between the motor nerve endings and the muscle.
● Because the amount of acetylcholine present in the synapse fluctuates, there is fatigability, which is the hallmark of myasthenia gravis: on repeated or sustained contraction, fewer receptors are available for activation and the strength of the muscle diminishes.
Clinical Presentation of Ocular Myasthenia
● Unilateral or bilateral fluctuating ptosis (▶Fig. 13.62) that may worsen in bright sunlight, perhaps because of an increased stimulus to blink
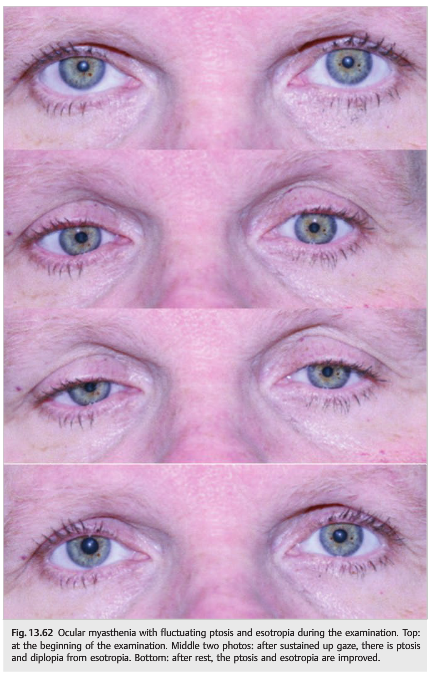
● Fluctuating binocular diplopia
● Symptoms and signs typically worsen after exercise or when patients are tired.
● Ptosis and extraocular movement fluctuate even during the examination.
● The symptoms and signs improve with rest (sleep test).
● The ptosis often improves when ice is applied on the ptotic eyelid (ice test).
● Ptosis is very common but may be absent (▶Fig. 13.63).
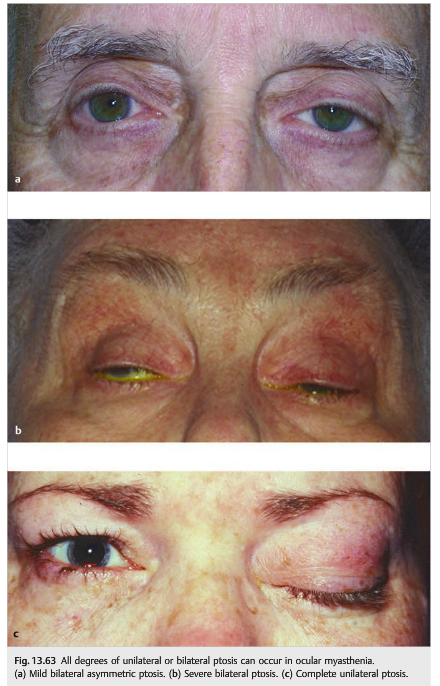
● Myasthenia can mimic any abnormal eye movement.
● The ophthalmoparesis can involve all the extraocular muscles or can be limited to one muscle or one group of muscles, thereby mimicking a pupil-sparing third nerve palsy, a sixth nerve palsy, or even an internuclear ophthalmoplegia. The neuromuscular junctions innervated by the third nerve are particularly susceptible to involvement by myasthenia.
● Diffuse, bilateral ophthalmoplegia with sparing of the pupil is highly suspicious for ocular myasthenia (▶Fig. 13.64).
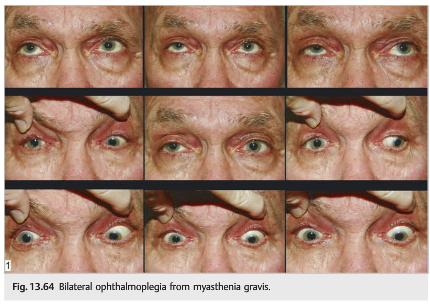
● The orbicularis muscles (used to close the eyes very tightly) are often also weak: it is then easy to force the eyes open.
● The pupils are always normal.
● Findings fluctuate from one examination to another and during the examination. After some rest, the ptosis and diplopia often resolve.
● Ocular myasthenia may be isolated or may precede (sometimes by a few months or years) or accompany systemic myasthenia.
● In systemic myasthenia, there can be proximal diffuse weakness of the extremities, change in voice (hoarseness or nasal quality), swallowing difficulties, and respiratory compromise. Systemic myasthenia is a medical emergency, particularly when respiration and swallowing are affected.
Pearls
Between 50 and 60% of patients presenting with purely ocular myasthenia will progress to develop generalized disease, and the vast majority of those who do will do so within 1 to 2 years. Generalized myasthenia gravis can be an emergency because patients may develop respiratory distress and swallowing difficulties.
In myasthenia gravis, the pupils are always normal and there is no pain.
Causes of Myasthenia Gravis
● Autoimmune disorder in most cases
● Rarely a paraneoplastic disorder
● There is an association in some patients between myasthenia and thymus enlargement or thymoma.
● Myasthenic syndrome may be induced by medications affecting neuromuscular transmission (curare, penicillamine). Autoimmune myasthenia gravis is often worsened by medications (curare, aminoglycosides, penicillamine, β-blockers, benzodiazepines).
Evaluation of the Patient with Suspected Ocular Myasthenia
The diagnosis of myasthenia is suspected clinically. A definite diagnosis is made when the clinical signs are reversed by the administration of a drug increasing the concentration of acetylcholine in the synapse (acetylcholinesterase inhibitor), when serum acetylcholine receptor antibodies are detected, or when there is electromyographic demonstration of muscle fatigability.
Examination and History
Examine the eyelids and eye movements.
● Are the signs fluctuating?
● Do the signs worsen with repetitive movements and sustained up gaze?
● Are the orbicularis muscles weak?
● Are the pupils normal?
Check for systemic involvement.
● Is there any proximal weakness?
○ Ask the patient to get up from a chair without using the hands 10 times in a row.
○ Ask the patient to raise both arms repetitively.
● Is the patient’s voice normal?
● Does the patient have any difficulty swallowing (coughs while eating or drinking)?
● Is the patient short of breath? ○ Ask the patient to count to 60 without breathing.
Determine the cause.
● List the patient’s medications.
● Any history of cancer?
● Any other autoimmune disease (such as thyroid disease)?
Confirm the diagnosis.
● Sleep test
● Ice test
● Administration of edrophonium (Tensilon test)
● Blood test for acetylcholine receptor antibodies (positive in up to 50% of patients with ocular myasthenia and in about 80 to 90% of patients with generalized myasthenia)
● Electromyography (single fiber electromyography is most accurate)
● Other tests
○ CT of the chest with contrast looking for a thymoma
○ Thyroid function tests
○ Autoantibody panel looking for other autoimmune diseases
Edrophonium (Tensilon) Test
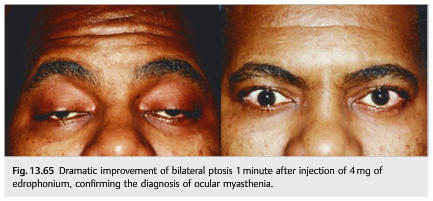
Edrophonium is a short-acting acetylcholinesterase inhibitor that increases the concentration of acetylcholine at the synapse and, therefore, temporarily reverses signs of myasthenia gravis. It is given intravenously and its effects last 2 to 3minutes. Side effects include sweating, fasciculations, diarrhea, abdominal cramps, palpitations, or bradycardia. Although it is generally well tolerated, hypotension, vagal reactions, and even respiratory or cardiac arrest are possible; it should be avoided in patients with known respiratory or cardiac diseases and in the elderly.
1. Identify an examination parameter to observe for improvement after edrophonium administration (ideally, ptosis).
2. Establish venous access.
3. Monitor blood pressure and heart rate.
4. Prepare edrophonium (10mg in 1mL syringe), 1 mg atropine, saline flush (10mL), blood pressure and heart rate monitoring equipment.
5. Inject 2 mg edrophonium test dose, flush intravenously (IV), observe for improvement or side effects for 1 minute. If definite improvement results, stop the test. You have a positive result.
6. Repeat step 4 every 1 to 2minutes with 2 mg edrophonium until improvement of the chosen parameter is obtained, side effects occur, or 10 mg of edrophonium is administered.
7. Alternatively, give a 5 mg edrophonium bolus after uneventful test dose and observe for improvement or side effects (▶Fig. 13.65).
Treatment of Myasthenia Gravis
The treatment of myasthenia gravis is mostly based on the clinical presentation and includes the following:
● Patients with generalized myasthenia should be evaluated emergently by a neurologist.
● Patients are instructed to go to the emergency room if they develop respiratory distress or swallowing difficulties.
● Medications associated with triggering or worsening of myasthenic symptoms and signs need to be discontinued (give the patient a list of forbidden medications).
● Treatment with pyridostigmine, a long-acting acetylcholinesterase inhibitor, is sometimes only partially effective for ophthalmoplegia.
● Corticosteroids (prednisone) are sometimes indicated if there is failure of pyridostigmine.
● Immunosuppressants are sometimes indicated for rebounds despite prednisone and as steroid-sparing agents.
● Thymectomy is indicated in all cases with a thymoma and in patients with severe generalized myasthenia.
● Symptomatic treatment of ptosis and diplopia: prisms and surgical procedures (on residual ptosis or strabismus) are sometimes necessary and should be performed in stable patients not responding to medical therapy. Ptosis crutches (mounted on glasses to keep the eyelids open) may be helpful.
Thyroid Eye Disease in Association with Myasthenia
Thyroid eye disease is the autoimmune disorder most commonly associated with ocular myasthenia. Each disease occurs more frequently in patients with the other disease than in the general population, attesting to the autoimmune origin of both disorders. Differentiating thyroid eye disease from myasthenia gravis can be challenging because both disorders present with painless bilateral ophthalmoplegia sparing the pupils.
Remember that thyroid eye disease produces proptosis, periorbital swelling, and lid retraction, whereas myasthenia produces ptosis. The presence of lid retraction in a patient with myasthenia raises the possibility of thyroid eye disease. The presence of ptosis in a patient with thyroid eye disease raises the possibility of myasthenia.
Myasthenia tends to preferentially affect those extraocular muscles innervated by branches of the third nerve (e.g., the levator palpebrae and the medial rectus), often manifesting with ptosis and exotropia, whereas thyroid eye disease preferentially affects the inferior and medial recti muscles, commonly manifesting with a restrictive hypotropia and esotropia.
When myasthenia and thyroid eye disease coexist, both diseases are treated simultaneously. Most patients respond to pyridostigmine and prednisone (▶Fig. 13.66).
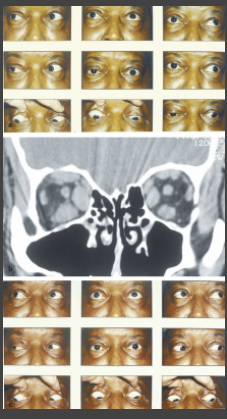
Fig. 13.66
(a) Diffuse bilateral ophthalmoplegia from ocular myasthenia and thyroid eye disease. Note the right ptosis, but also periorbital swelling and left lid retraction. There is mild bilateral proptosis.
(b) Coronal computed tomographic scan of the orbits without contrast showing enlarged extraocular muscles (right worse than left) consistent with thyroid eye disease.
(c) Same patient after treatment. The ptosis has resolved and the extraocular movements are full.
Reference: 1. Neuro-ophthalmology Illustrated-2nd Edition. Biousse V and Newman NJ. 2012. Theme
These questions are archived at https://neuro-ophthalmology.stanford.edu
Follow https://twitter.com/NeuroOphthQandA to be notified of new neuro-ophthalmology questions of the week.
Please send feedback, questions, and corrections totcooper@stanford.edu.